- Description of Services
- Scientific Review Committee
- Data Safety Monitoring Board (DSMB)
- Internal Monitoring and Auditing
- Sponsor Monitoring and Auditing for Clinical Research Studies
- Remote and On-Site Monitoring FAQs
- Preparing for an FDA or Sponsor Inspection
- ClinicalTrials.gov
- Human Gene and Cell Therapy Program
- Regulatory Consultations
- FDA Regulatory Affairs
- Contact Us
Description of Services
The Office of Regulatory Affairs (ORA) provides a broad spectrum of support for Clinical Investigators and their study teams in the conduct and navigation of clinical research regulatory requirements.
Services provided by this office include: Scientific and Feasibility Review, Data and Safety Monitoring, internal monitoring and auditing support, FDA and Sponsor inspection/audit preparation and guidance, ClinicalTrials.gov registration and results reporting assistance, FDA IND/IDE guidance and support, regulatory binder preparation, and more.
The mission of the ORA is to guide and support the UCLA clinical research community through the different compliance requirements associated with the conduct of clinical research.
Scientific Review Committee
Established in September 2016 and formally mandated in July 2022, the UCLA Scientific Review Committee (SRC) provides a scientific and feasibility review for non-oncology studies that meet the National Institutes of Health (NIH) definition of a Clinical Trial and that have not already been reviewed by an external scientific review committee. This scientific review is intended to complement the Institutional Review Board (IRB) review through a detailed review of the required elements of the clinical protocol, statistical applications, adequacy of research staffing, any competing trials, well-constituted data collection forms, and utilization of institutional resources.
The Committee meets on the first and third Wednesday of every month. Meeting Schedule with deadlines.
Investigators are not required to initiate SRC reviews and there is no application process. All study documentation is collected by the ORA staff and provided to the SRC for review. SRC review occurs prior to IRB review.
Please email the UCLA SRC with any questions.
Please see SRC policy 916 for more information.
Please see Scientific Review FAQs for more information.
Data Safety Monitoring Board (DSMB)
A Data and Safety Monitoring Board (DSMB) is a group of individuals with pertinent expertise that reviews accumulating data from an ongoing clinical trial. The CTSI DSMB offers oversight for those investigator initiated trials that do not have an external DSMB oversight mechanism. The DSMB advises investigators regarding the continuing safety of trial subjects and those yet to be recruited to the trial, as well as the continuing validity and scientific merit of the trial.
The CTSI DSMB performs the following general functions:
- Objectively appraise a study’s progress
- Assess data quality via a formal and planned process
- Provide analytical expertise and rigor
- Determine the statistical significance of efficacy and/or risk‐benefit ratio
Required forms for submission to DSMB to be submitted to the Office of Regulatory Affairs.
Learn more about Data & Safety Monitoring at UCLA
Internal Monitoring and Auditing
Sponsor-Investigators are responsible for the selection of qualified study monitors and ensuring that the trials are adequately monitored throughout the life of the trial. At UCLA, the Office of Regulatory Affairs offers assistance with monitoring and quality assurance auditing for investigator-initiated studies. This service helps ensure compliance with FDA, GCP, and IRB regulations, and UCLA Health System policies and guidance, as related to clinical research. Please contact the Office of Regulatory Affairs for more informaton.
The ORA monitoring program provides a proactive (rather than “for cause”) regulatory assessment and has a strong educational component. Investigators are required to provide monitoring findings to the IRB according to their policies.
The ORA auditing program includes routine and for-cause reviews (requested by institutional officials). The purpose of routine reviews is to assist investigators with achieving and maintaining regulatory compliance. The reviews are meant to be educational rather than punitive in nature. The ORA summarizes and reports the findings directly to the investigators and the CTSI DSMB.
When writing a grant proposal, Investigators are encouraged to include costs for monitoring and auditing of their study. Contact the Office of Regulatory Affairs for details.
Sponsor Monitoring and Auditing for Clinical Research Studies
- Remote monitoring through HealthLink (a module within UCLA’s CareConnect Electronic Health Records System) for CareConnect related source records, eBinders for regulatory documents, and UCLA’s secure instance of Box.com for source documents not stored in CareConnect;
- Remote monitoring through UCLA’s secured instance of Zoom video conferencing facilitated by the principal investigator and research team; and/or
- Safe and compliant on-site monitoring facilitated by the principal investigator & research team as outlined by the following procedures and/or policies (links may require UCLA AD login):
Remote monitoring through HealthLink requires both, a Remote Monitoring Agreement facilitated by Clinical Trial Contracting & Strategic Relations (CTC-SR), as well as budget allocations for applicable Remote Monitoring Set-Up and Provisioning Fees (referenced below) for each sponsor-monitor provisioned with remote monitoring access. The institutional remote monitoring agreement and applicable remote monitoring fees have been standardized to streamline remote monitoring setup and mitigate negotiation. The option for remote monitoring may be made available prospectively during contract negotiations for new clinical trial agreements received by CTC-SR as of October 1, 2020. For existing studies, a process has been established to prioritize study teams and sponsors requiring remote monitoring access with the limited resources available to support remote monitor contracting, set-up and provisioning. For more information, please contact CTSIORA@mednet.ucla.edu.
Remote monitoring of regulatory documents through eBinders requires budget allocations for applicable Remote Monitoring Set-Up and Provisioning Fees (referenced below) for each sponsor-monitor provisioned with remote monitoring access. The remote monitoring fees have been standardized to streamline remote monitoring setup and mitigate negotiation.
For source and study documents stored external to CareConnect, such documents should be redacted appropriately of participant Protected Health Information (PHI) and uploaded to UCLA secure Box.
On-site Monitoring has traditionally been the primary method for sponsor monitoring of clinical trials at UCLA. However, on-site monitoring should be limited to only rare instances when the remote monitoring arrangements referenced in (1) and (2) above cannot be achieved with sponsor(s).
- On-site monitoring can occur in Non-UCLA Health System Space (Departmental or School) where the on-site monitoring access is deemed essential to comply with applicable laws and study teams and monitors strictly adhere to the appropriate UCLA health visitor requirements. Investigators must obtain Department Chair or Division Chief (or designee) written approval permitting the request for an on-site visit.
- On-site monitoring can occur in UCLA Health System Space where the on-site monitoring access is deemed essential to comply with applicable laws and study teams and monitors strictly adhere to the appropriate UCLA Health vendor requirements. For visits in Health System Space, the study Principal Investigator must obtain Department Chair or Division Chief (or designee) written approval permitting the request for an on-site visit. In addition, approval must be obtained by contacting the CTSI Office of Regulatory Affairs. The monitor will need SEC³URE GO! access to enter UCLA Health System Space for their monitoring visit.
- "UCLA Health System Space" is any location owned or operated by UCLA hospitals and/or UCLA outpatient clinics, managed by either the UCLA Hospital or UCLA Faculty Practice Group utilized to provide clinical care to UCLA patients. This includes, but is not limited to, Ronald Reagan UCLA Medical Center, Santa Monica Medical Center, UCLA Orthopaedic Surgery, Resnick Neuropsychiatric Hospital, ambulatory sites, and the UCLA Faculty Practice Group.
For research studies where a study sponsor is NOT obligated by the FDA to monitor/audit the study source records, sponsors are generally not allowed to conduct monitoring of study records. Sponsors may request a special exception with the Office of Compliance. If approved, sponsors may only conduct monitoring through the remote access process.
|
Remote Monitoring Fee Schedule |
Remote Monitoring EHR Setup Fee (includes study setup and provisioning up to one monitor for both HealthLink and eBinders) |
Remote Monitor EHR Setup Fee (each additional monitor) This covers both HealthLink and eBinders) |
eBinders Only Remote Monitoring Setup Fee (includes study setup and provisioning up to one monitor for only eBinders) |
eBinder Only Remote Monitor Setup Fee (each additional monitor) This covers only eBinders |
Non-EHR, cloud-based remote monitoring setup (e.g. participant diaries, questionnaires, etc.) |
Remote or On-Site Monitoring Visit Fee (applicable if UCLA faculty and/or staff visit participation is required) |
|---|---|---|---|---|---|---|
|
Fee Classification |
Required Pass-Thru |
Required Pass-Thru |
Required Pass-Thru |
Required Pass-Thru |
Guidance for Study Team Effort |
Guidance for Study Team Effort |
|
Per Study, Per Monitor, Per Visit |
Per Study |
Per Monitor |
Per Study |
Per Monitor |
Per Study |
Per Monitoring Visit |
|
Industry Funded Clinical Research Study |
$3,000 |
$2,250 |
$1,000 |
$500 |
$1,000-$2,500 |
$250-$500 |
"Please note: all fees above are subject to the applicable UCLA indirect rate(s)."
These rates apply to all NEW studies initiated within webIRB and BruinIRB as of March 1, 2024. For prior rates, please contact the CTSI ORA.
- Remote monitoring terms and obligations contract review and execution with study sponsor(s) and CRO(s) for each applicable clinical research study.
- Study-specific statement of work defining scope and effective timeline for provisioning.
- Individual study monitor terms and obligations agreement review and completion.
- UCLA Healthlink Electronic Health Record (EHR) user access application review and completion.
- Remote study monitor provisional access application completion.
- Remote study monitor online training – scheduling, facilitation, and completion.
- Remote study monitor approval and access provisioning.
- Remote study monitor virtual visit scheduling and research participant linking to Healthlink.
- Remote monitoring use and access compliance – tracking, auditing, maintenance and reporting.
Monitors may not in any way divulge, save, copy, print, record, photograph, download, export, screenshot, release, sell, loan, alter and/or destroy any PHI except as permitted by law and properly authorized by the policies of the Participant. Any breach of the responsibilities and/or conditions of the terms of access may be subject to access suspension, employer notification and disciplinary action, and may be subject to civil and/or criminal charges, as applicable. Reinstatement of suspended access will be subject to the requirements, training and fees associated with new user access, as determined at UCLA’s discretion.
Please see the “UCLA Clinical Research External Monitoring & Auditing Policy HS 9207” for more information.
For access to HealthLink and eBinders, once the Remote Monitoring Agreement has been executed with Clinical Trial Contracting & Strategic Relations (CTC-SR), please complete the following forms and submit them to the CTSI ORA:
- Monitor HealthLink Request Form
- User Agreement - UCLA Health EHR Access Agreement
- Confidentiality Statement
- Instructions for Completing Monitoring Forms
For access to eBinders ONLY, please complete the following forms and submit them to the CTSI ORA:
- Monitor eBinders Request Form
- eBinders Training Certificate
*eBinders coming Spring 2024
Remote Monitoring
- What is the remote monitoring set up fee for the combination of HealthLink and eBinders?
For Industry (for profit) funded studies the cost is $3000 per study. The $3000 cost covers the provisioning of a study and one monitor for that study. For each additional monitor to be provisioned for that study, it is an additional $2250. - Is the $3000 a one-time fee, or is it per remote monitoring visit (RMV)?
Yes, the $3000 is a one-time fee. It is the study set up fee and also covers provisioning one monitor for that study. For each additional monitor to be provisioned for that study, it is an additional $2250. An additional $2250 will be charged each time a new monitor comes onto the study. The cost is not per RMV. If there is staff time on your end, you will need to discuss those charges separately. - What is the remote monitoring set up fee for only eBinders?
For Industry (for profit) funded studies the cost is $1000 per study. The $1000 cost covers the provisioning of a study and one monitor for that study. For each additional monitor to be provisioned for that study, it is an additional $500. -
We have already been provisioned and paid for access to HealthLink, do we need to pay an extra fee to now access eBinders?
No, it is one fee for access to both systems. - Are the signed forms (HealthLink user access form, confidentiality statement form, eBinders user agreement, and Individual user access form) a one-time requirement or do they need to be provided prior to each RMV?
The signed forms are a one-time requirement only. - Our monitors have already been provisioned for HealthLink, how do we give them access to eBinders?
For access to eBinders ONLY, please complete the following forms and submit them to the CTSI ORA:- Monitor eBinders Request Form
- eBinders Training Certificate
- If a monitor has already been provisioned for a study (eg. study A) and is now requesting access to another study (study B), are they required to provide the signed forms again?
No, it is not required. The study team/Sponsor/CRO contact should e-mail the CTSI ORA at ctsiora@mednet.ucla.edu requesting access for the monitor for the additional study and provide the following information:- i. Name of the previously provisioned monitor
- ii. IRB# for the study they are requesting access to
- If the study has a fully executed remote monitoring agreement, the provisioning process will be initiated immediately.
- Do clinical research coordinators need to be trained to provide access to specific patients during the specified time frame?
Once the CRAs EMR and eBinders access is provisioned, the study team will be provided with a tip sheet that the clinical coordinators can refer to, for help releasing subjects in HealthLink and study documents in eBinders. - Who on the study team can release subject records to CRAs for remote review?
Only certain study personnel like research nurses or clinical research coordinators are able to release subject records. Data managers are not able to do so. The system has been set up in this manner based on a study personnel’s job description. - Will monitors only have access to the subject records they are assigned to review or all the subjects. For example, if monitor A is assigned to review subjects 001 and 002 on a study and Monitor B is assigned to review subjects 003 and 004 on the same study, will both monitors have access to all 4 subjects on the study?
Yes. Coordinators release patients to a specific HL (Healthlink) patient list (the Patient Group), which is built by study. The research monitors are given access to specific studies (the User Context, which is linked to the Patient Group). A research monitor who has been provisioned with a User Context for a specific study will have access to all the patients who have been released to the study. In this scenario, if a monitor should not access specific patient charts, this should be a training point to instruct the monitor to not open the charts they should not open. Every access is fully auditable, so if anything ever gets called into question there is an audit trail that the Healthlink team can look back on. - How long is each remote monitoring visit (RMV)?
Each RMV is capped at 5 days. The system will generate an error message if the coordinator tries to release subject records for longer than 5 consecutive days. After 5 days (or shorter if set for less time by the study team), the subject records will no longer be able to be seen by the monitor. This ensures the CRAs’ access is limited to the dates specified for the RMV. - Is there a way to undo release of a subject for monitoring? For example, if we learn that a monitor doesn’t need to review screen fails, how would we remove their access from those patients, so that we can minimize PHI access?
Yes, if you need any records unreleased prior to the expiration (duration when remote access to the subject records is available to the CRA) please e-mail the CTSI ORA at ctsiora@mednet.ucla.edu. - How long is the EMR and e-regulatory access valid?
Once provisioned, the monitor will have approved access to the system for up to one year, that can be renewed annually for the life of the study or up until the monitor is no longer associated with the study, whichever happens first. - How often can monitors schedule these RMVs?
Monitors can schedule RMV as many times as necessary depending on the study teams' availability. Once the monitor has access to the study, they should communicate with the study team to schedule RMVs. The study team connects the subject records to the study monitor and sets the remote review dates based on the mutually agreed upon remote monitoring dates. - Once approved to remote monitor, how would monitors go about scheduling these RMVs?
The local team would handle the scheduling of the monitoring and connecting of subjects with the monitoring visit. Once the CRA has EMR access, they should reach out to the study team to schedule remote monitoring visits. Once they mutually agree to the dates, the study personnel authorized to release subject records will go into the system to release subject records for that particular remote visit. Subject records may be released for review for up to a max of 5 days. If the remote visit is only scheduled for 2 days, the release of records can be restricted to only those 2 days or as many days as the visit is scheduled for, but may not exceed 5 consecutive days. The system will generate an error message when attempting to release records beyond 5 days. - Once in-person monitoring visits are re-instated, will CRAs lose access to the remote EMR?
There are no current plans to stop remote visits. - What does the HealthLink monitoring training for CRAs entail?
It is an online training for CRAs on navigating HealthLink. It is pretty quick and easy to complete. The study team will be provided with a tip sheet that they can use to link the subject to the RMV and release records to the CRA for remote review. - What does the eBinders monitoring training for CRAs entail?
It is an online training for CRAs on navigating eBinders. It is quick and easy to complete. The study team will be provided with a tip sheet that they can use to release study documents to the CRA for remote review. - Does the Remote monitoring access include the use of Box or any other systems to share files that wouldn’t normally be in CareConnect (such as questionnaires or regulatory files)?
Documents that are not in CareConnect can be redacted for subject identifying information and uploaded to the UCLA Health Box system. You must limit the monitor’s view to read only. The ORA office is not involved in this process. If there is staff time on your end for setting this up, you will need to discuss those charges separately with the sponsor. - If a monitor has already been provisioned for a study (eg. study A) and is now requesting access to another study (study B), are they are required to provide the signed forms again?
Additional signed forms are not needed. However, the $2250 additional monitor fee will apply to this type of request. - Do the additional monitor fees still apply to CRAs that are being replaced/no longer with the company?
Yes, the fee applies even if the CRA is being replaced for the study. -
How long does the remote monitoring provisioning process take?
The process typically takes 8-12 business days once the remote monitoring agreement has been executed.
*eBinders coming Spring 2024
On-Site Monitoring
Health System Space
-
What is SEC³URE GO!?
SEC³URE GO! is a wearable, digital badge that combines mobile check-in with visitor identification. It visually displays the user’s compliance status for all facility staff to see. - Who can use SEC³URE GO!?
Any commercial and clinical visitors to your facility can use it, including sponsor monitors, clinical contractors, physicians, and nurses. However, they will need a current SEC³URE Passport to be eligible for the badge. - What does the SEC³URE GO! check-in process look like for the monitor?
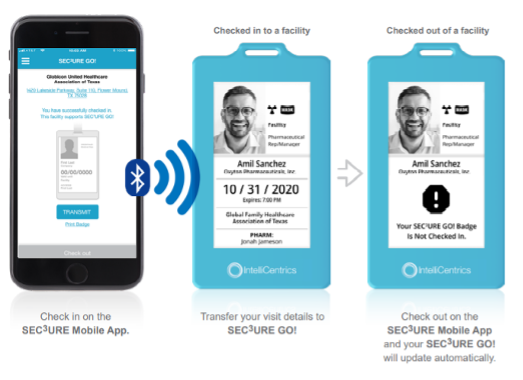
- I didn’t receive the email to set up my login to the SEC³URE GO! platform. What do I do?
Contact customer service at 817-SEC3URE or 817-732-3873 - Additional monitors need access to the SEC³URE GO! platform. How do I get them set up?
Contact customer service at 817-SEC3URE or 817-732-3873 - What is the SEC³URE GO! cost per monitor?
$334/year - If a monitor works at more than one facility, do they have to register at all locations, or does one registration cover all locations?
One registration covers all locations. - What is the Contact/Support Information?
Customer service – 817-SEC3URE or 817-732-3873 - What are the standards and requirements for all users:
Non-Health System Space
- What are the links for non-health system requirements for visitors?
Preparing for an FDA or Sponsor Inspection
Upon notification of an FDA inspection, please contact the Office of Regulatory Affairs immediately for guidance and assistance. The ORA provides one-on-one inspection/audit preparation guidance, education on how to interact with the FDA, and provides support for responding to the FDA’s findings, if needed.
If there is a concern about the study preparedness for a Sponsor audit, contact the Office of Regulatory Affairs to request an audit readiness assessment for both industry and investigator-initiated studies. This program helps ensure compliance with FDA, GCP, and IRB regulations, and UCLA Health System policies and guidance, as related to clinical research. The results of the pre-audit assessment will be provided for investigators and teams.
Visit FDA Inspections & Alerts to learn more.
ClinicalTrials.gov Registration and Reporting
ClinicalTrials.gov is a website and online database of clinical research studies and information about their results. The purpose of ClinicalTrials.gov is to provide information about clinical research studies to the public, researchers, and health care professionals. The U.S. government does not review or approve the safety and science of all studies listed on this website.
ClinicalTrials.gov:
- Relies on sponsors or investigators to submit and update information about studies
- Lists up-to-date information on clinical research studies and their results with new studies added almost every day
- Includes studies that take place in all 50 states and over 200 countries
- Supports laws, regulations, and policies that require sponsors and investigators to publicly share information about clinical trials, including results
Most interventional studies with health outcomes must be registered, and may be required to report results, in ClinicalTrials.gov. Those responsible for conducting a clinical trial must ensure that they are in compliance with these requirements for:
- All NIH-funded trials including phase 1 studies and clinical trials of behavioral or non-FDA-regulated interventions (Registration and Results required)
- Clinical trials involving FDA-regulated drug, biologic and device products (Registration and Results required).
- Studies that will bill routine costs to Medicare or any other insurer (Registration required)
- Clinical trials intended for publication in a journal recognized by the ICMJE (Registration required).
- Informed Consent Statement
Registration may be required by law and/or policy if any one (or more) of the following is true:
Required by Your Funding Source
Required for Journal Publication
The CTSI Office of Regulatory Affairs (ORA) offers assistance to UCLA investigators for registration and/or results reporting of investigator-initiated clinical trials (IITs):
- ClinicalTrials.gov Protocol Registration: ORA to perform registration entries in the Protocol Registration System (PRS) for ClinicalTrials.gov in compliance with Protocol Registration Quality Control Review Criteria https://prsinfo.clinicaltrials.gov/ProtocolDetailedReviewItems.pdf, and update/verification as needed through study duration (minimum annually)
- ClinicalTrials.gov Results Reporting – Option #1: ORA will provide fillable tables or lists specifying results data required, to be completed and returned via email, and ORA will perform data entry. ORA will respond to reviewers’ comments, with information provided by PI/statistician as needed.
- ClinicalTrials.gov Results Reporting – Option #2: ORA will meet with PI/statistician via one or two ZOOM meetings to (1) explain PRS data entry requirements for results reporting, (2) guide through data entry process for results reporting and statistical analyses (if applicable), (3) Review and provide feedback prior to submission on entries likely to violate review criteria.
|
Registration and Results Reporting |
ClinicalTrials.gov Protocol Registration |
ClinicalTrials.gov Results Reporting – Option #1 |
ClinicalTrials.gov Results Reporting – Option #2 |
|---|---|---|---|
|
Fee Classification |
Required Pass-Thru |
Required Pass-Thru |
Required Pass-Thru |
|
Per Study, Per Monitor, Per Visit |
Per Study |
Per Study |
Per Study |
|
Fee |
$1,500 |
$2,500 |
$1,000 |
If you have questions or need assistance, please contact the CTSI Office of Regulatory Affairs.
Human Gene and Cell Therapy Program at UCLA
The UCLA Human Gene and Cell Therapy Program (HGCTP) is a collaborative effort of the David Geffen School of Medicine, the Jonsson Comprehensive Cancer Center, and the Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research. The HGCTP provides scientific, safety, and quality assurance oversight of all Human Gene Transfer Clinical Trials conducted at UCLA and supports a manufacturing facility for cell and gene transfer products. More details are available in the HGCTP policy.
Approval by Institutional Bodies
All UCLA Human Gene Transfer research must be approved by the following institutional bodies before work can begin:
- Institutional Review Board (IRB)
- Jonsson Comprehensive Cancer Center (JCCC) Internal Scientific Peer Review Committee (ISPRC) for oncology studies or UCLA Scientific Review Committee (SRC) for non-oncology studies
- Institutional Biosafety Committee (IBC) (See UCLA Policy 992 and IBC Requirements for Human Gene Transfer (HGT) Studies)
- Medical Radiation Safety Committee (MRSC), if applicable
Scientific and Quality Assurance Oversight
Scientific review, data, and safety monitoring and quality assurance must be completed.
- The JCCC ISPRC for oncology clinical trials and the UCLA SRC for non-oncology clinical trials, will review the clinical protocol, statistical plan, and other factors such as adequate staffing, competing trials, etc.
- In addition to scientific integrity, the committees also review data and safety monitoring plans for the studies and if necessary, recommend trial oversight by their respective data and safety monitoring board (DSMB).
- All studies overseen by an institutional DSMB are subject to monitoring and auditing by the quality assurance officers within either the JCCC or the Clinical and Translational Science Institute (CTSI).
For more information regarding scientific review, DSMB services and quality assurance oversight, please see the JCCC ISPRC website, the JCCC DSMB website and the CTSI ORA website.
Required Training
All UCLA Key Personnel involved with Human Gene Transfer Clinical Trials must have completed and be up to date with the following courses offered through the Collaborative Institutional Training Initiative (CITI) program:
- Biomedical Good Clinical Practice Training (See UCLA Policy 917);
- NIH Recombinant DNA Guidelines*; and
- Human Gene Transfer*.
*Training is valid for 3 years.
Manufacturing of Gene Transfer Products
The UCLA Human Gene and Cell Therapy Facility (HGCTF) supports manufacturing of gene and cell therapy products for UCLA Principal Investigators as well as other academic and industry partners conducting Clinical Trials in which a cell or gene therapy product is manufactured under an FDA IND. For more information, see the HGCTF website: https://medschool.ucla.edu/research/human-gene-and-cell-therapy-facility
Contact Information
- For non-oncology studies, contact the Clinical and Translational Science Institute (CTSI) Office of Regulatory Affairs (ORA): ctsiora@mednet.ucla.edu
- For oncology-related studies, contact the Jonsson Comprehensive Cancer Center (JCCC) Internal Scientific Peer Review Committee (ISPRC): CORA@mednet.ucla.edu
- For questions related to manufacturing a cell or gene transfer product, please contact the Human Gene and Cell Therapy Facility: GMP@mednet.ucla.edu
- For questions about Institutional Review Board (IRB) review and approval, contact the MIRB at mirb@research.ucla.edu
- For questions about Institutional Biosafety Committee (IBC) review and approval, contact the IBC administrative staff: ibc@research.ucla.edu
- For questions about the Medical Radiation Safety Committee, contact the RSC administrative staff: rsc@research.ucla.edu
HGCTP and UCLA Oversight Committees

*Oversight includes:
1. Scientific Review of Protocols
2. Data and Safety Monitoring
3. Quality Assurance (Monitoring and Auditing of Trial Conduct)
4. Training
Regulatory Consultations
The Office of Regulatory Affairs offers a wide variety of regulatory consultations to Clinical Investigators and their study teams in the navigation of the regulatory process.
ClinicalTrials.gov
The Food and Drug Administration Amendments Act of 2007, Section 801 (FDAAA 801) requires Responsible Parties to register and submit summary results of clinical trials with ClinicalTrials.gov and applies to certain Clinical Trials of drugs (including biological products) and medical devices. The International Committee of Medical Journal Editors (ICMJE) requires trial registration as a condition of the publication of research results generated by a clinical trial as required by ICMJE. Finally, Centers for Medicare & Medicaid Services (CMS) require inclusion of an 8-digit Clinical Trial number from ClinicalTrials.gov on claims associated with Clinical Trial participation.
The ORA provides support, for non-cancer studies, to assist and advise Principal Investigators with their obligations. Please contact Elaine Cooperstein for guidance on registration, results reporting, and a PRS account.
Regulatory Binder Preparation
A Regulatory Binder assists sites in achieving and maintaining regulatory compliance and ensuring the highest standards of human subject research. Regulatory binders house all study documentation including, but not limited to, the study protocol, staff CVs, licenses, logs, IRB documents, consent forms, data collection/CRFs, lab documents, sponsor documents, drug/device accountability, FDA documentation, financial disclosure documentation, DSMB information, and more.
For guidance on developing a regulatory binder or evaluation of your current binder, please contact Associate Director, Uma Ganapati PhD.
FDA IND/IDE Guidance and Support
Support for investigators holding an IND or IDE at all stages of an investigation including:
- Determination of product classification (i.e., drug, device, combination product, biologic)
- Applicability of an IND or IDE
- Assistance with IND or IDE application and subsequent submissions (amendments, safety reports, annual and final reports)
- Preparation, coordination, facilitation, and attendance at FDA meetings
- Preparation for and regulatory support during FDA inspections of investigator-sponsored clinical trials
- Update regarding new guidance documents, inspection trends, inspection actions and new regulatory actions taken by FDA relating to clinical trials
Please contact Director of FDA Affairs, Marlene Berro MS, RAC for additional information.
More
Individual and small group trainings and lectures covering good clinical practice and the conduct of clinical research. To request a training or for other clinical trial regulatory affairs questions, please contact us.
FDA Affairs
Located in the CTSI Office of Clinical Research, the FDA Affairs team provides FDA support and guidance for investigators submitting or holding an IND or IDE at all stages of an investigation. In addition, the FDA Affairs team has created a virtual clinical research platform called ResearchGo that provides a single portal to a wealth of resources, expertise, and best practices for investigators and research staff to facilitate efficient, compliant and ethical study conduct and management.
What We Do
- Determination of product classification (e.g., drug, device, combination product, biologic)
- Applicability of an IND or IDE
- Assistance with IND or IDE application and subsequent submissions (e.g., amendments, safety reports, annual and final reports)
- Preparation, coordination, facilitation, and attendance at FDA meetings
- Preparation for and regulatory support during FDA inspections of investigator-sponsored clinical trials
- Update UCLA research community regarding new guidance documents, inspection trends, inspection actions and new regulatory actions taken by FDA relating to clinical trials
- Drug, Device, and Biologic Protocol Development
- Consult with TDG and other campus entities including UCLA Anderson School and School of Bioengineering regarding Drug and Device Development
Who We Are
Marlene Berro, MS, RAC - Director, FDA Affairs
Jenny Ahn, BSN, RN - FDA Specialist
Amanda Gonzales, MPH, CCRP - FDA Specialist
Contact Us
Terra Hughes, M.S., Director, CTSI Office of Regulatory Affairs
- Scientific Review Committee
- Data and Safety Monitoring Board
- Training and Lectures
- General Questions
Uma Ganapati, Ph.D., Associate Director, CTSI Office of Regulatory Affairs
- Internal Auditing and Monitoring
- FDA and Sponsor Inspection/Audit Preparation
- Regulatory Binder Preparation
Elaine Cooperstein, MS, CCRP, ClinicalTrials.gov Liaison, CTSI Office of Regulatory Affairs
- ClinicalTrials.gov, including PRS account access, registration, and resulting reporting
Marlene Berro, MS, RAC, Director, FDA Affairs
- FDA IND/IDE Guidance
- ResearchGo Site
Required forms for submission to DSMB to be submitted to the Office of Regulatory Affairs.